Detect 5x innovation signals
Save time
Better internal reporting & communication
already trust us
Documents
Experts
Organizations
Topics
With InnoScout, our tech scouting tool, you can do more than search.
Analyze massive amounts of scientific and industrial data sources in a few steps discovering keywords relevant in your field of interest, and get updated insights on emerging technologies and innovative companies and startups worldwide.
Book a Demo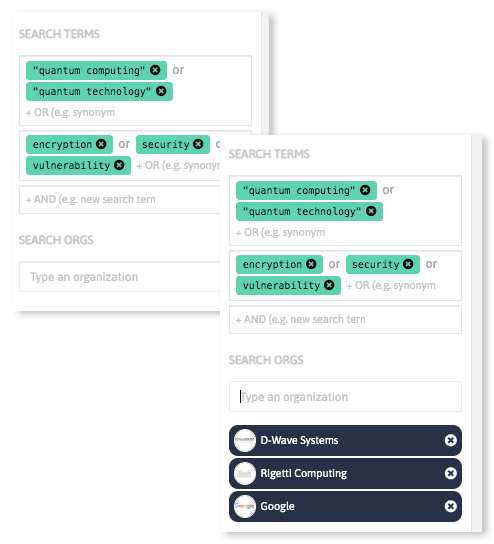
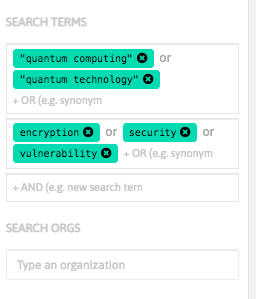
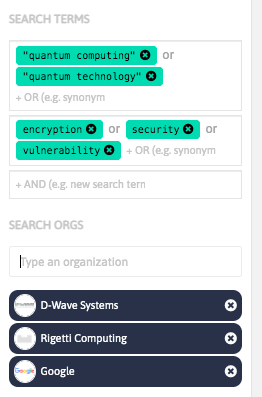
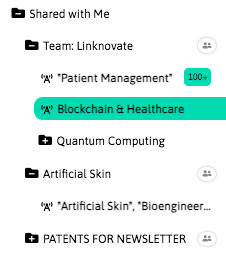
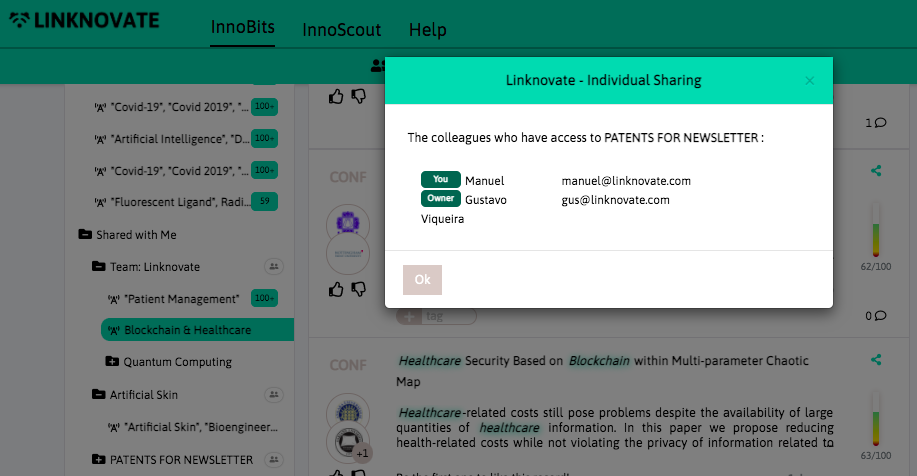
With InnoBits, our monitoring system, you can save searches and get real-time updates on the latest trends, patents, scientific publications, and news about specific sectors. Decide who you share it with, customize your search results based on your preferences.
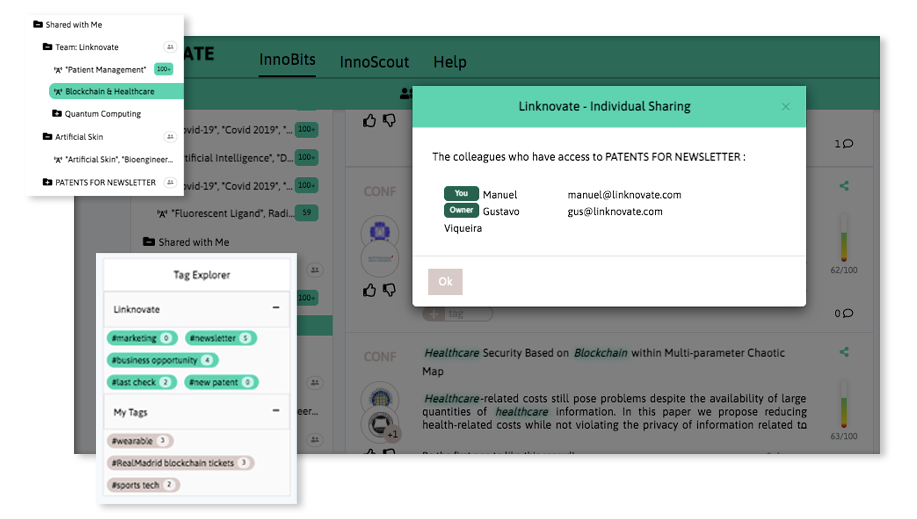
Share your discoveries with your team and collaborate with them by creating multiple searches into your internal innovation observatory. Joint team-based scouting is stronger than single-handed research.
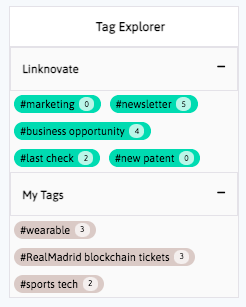
Book a DemoDistill and report what matters, facilitate business innovation. We help you communicate internally, download graphs, export results and insights, and generate reports and newsletters in a few clicks.


“Through Linknovate I have obtained info about experts and references from start-ups and SMEs with activity in the field under study, in addition to accessing cross-information: relevant connections between academic and business profiles.”
Senior Project Lead & Technical Advisor
“We can monitor and follow relevant information on many different types of innovation and tech news conveniently and quickly in one platform, no matter the topic or specific industry”
Partenariat, Startups & RI Responsable
“We use both InnoScout and Innobits functions in our innovation team. Both features combined are a very convenient way to scout and monitor markets and technologies.”
Senior Lead Innovation & New Business
"The usability of the platform and how it helps us to narrow down our search to very specific topics is what adds most value to our scouting teams right now.”
Management Innovation Manager

"The Innobits tool within the Linknovate platform helps us to monitor the clinical horizon. Not only can we quickly scan a broad range of sources and content items, the platform also allows us to organise and store insights and collaborate well as a team. Key to this is the platform’s functionality and willingness of the Linknovate team to continually adapt and innovate on their platform to meet the needs of their client base.”
Technology Observatory of Global Healthcare Provider
We founded Linknovate in 2012 at Stanford University accelerator program (StartX), CA.
It has been an amazing journey, we invite you to be a part of it (and if you wish, to visit
us
in Galicia, Spain).

Stanford University Accelerator Alumni

1st Prize. European Open Data Competition

Berlin Program Alumni

Multiple Grant Awardee